





“在内分泌疾病中,有一种疾病被学者形象地描述为“the great mimickers”, “the great masquerader”和“Occult”,意为“模仿者”、“冒充者”和“神秘的疾病”。这就是今天要和大家扒一扒的“嗜铬细胞瘤”。

历史回顾
1886年Felix Fräkel描述了一例18岁恶性高血压女患者Fraulein Minna Roll,在尸检中证实其肾上腺占位。1896年病理学家Manasse发现铬盐可使发生于肾上腺髓质的肿瘤细胞染色为深棕色。1912年病理学家Pick利用希腊文的黑色(phaios)、颜色(chroma)和细胞(kytos)3个词合成创造新词pheochromocytoma(嗜铬细胞瘤),被一直沿用至今。1939年北京协和医院诊断国内第一例PCCs。自上世纪90年代以来,学术界对本病的关注逐渐由临床表型联系到了基因型。(见 图1)
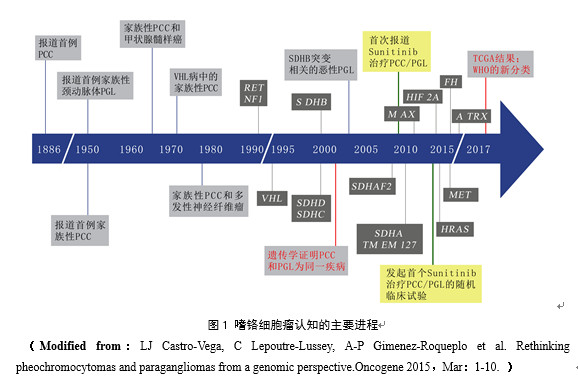

分类
嗜铬细胞瘤除可见于肾上腺髓质外,亦可发生在自主神经系统中枢及其支配神经(从颅底至骨盆底均有分布)或身体其他部位嗜铬组织(见图2)。
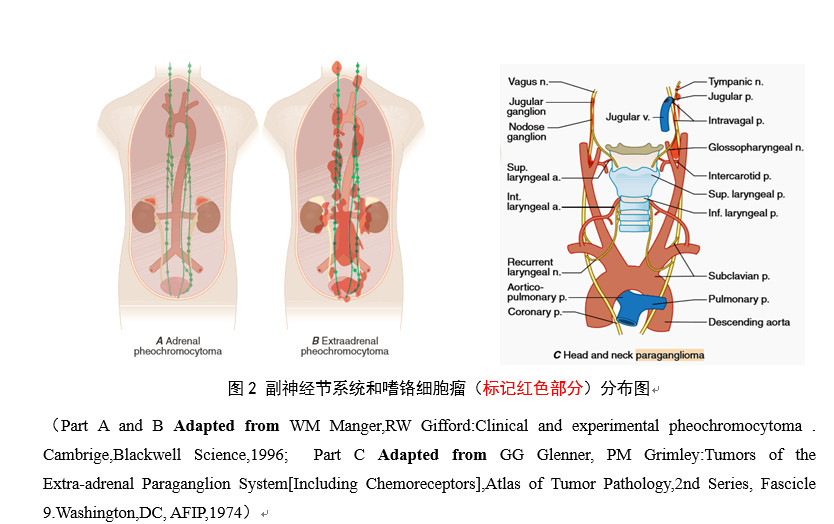
2017年世界卫生组织(WHO)将肾上腺髓质和肾上腺外副神经节瘤分为肾上腺嗜铬细胞瘤(Pheochromocytoma,PCC)、肾上腺外副神经节瘤(Paragangliomas,PGLs)、肾上腺神经母细胞瘤、复合型PCC和复合型PGL。其中,肾上腺外PGLs又分为头颈部PGLs(HNPGLs)和交感神经PGLs。习惯将PCCs和PGLs合称为PPGLs。
既往曾对PPGLs分良恶性,但2017年WHO肾上腺肿瘤分类认为,所有PPGLs均有转移潜能,故用“转移性”替代“恶性”的诊断。

流行病学及病因
嗜铬细胞瘤是人类罕见的神经内分泌肿瘤,每年每百万人口中仅约0.4-9.5人发病,约占所有高血压患者的0.1%。从上世纪90年代,人们逐步发现,RET、VHL、NF1、SDHB、SDHC、SDHA、SDHAF2、TMEM127和MAX等基因的胚系和/或体系突变引起本病的发生。而且,超过1/3的PPGLs与遗传性肿瘤易感综合征有关, 如:1型神经纤维瘤病(NF1)、多发性内分泌腺瘤病2型(MEN 2)、von Hippel-Lindau综合征(VHL)、PGL1~5型、Carney-Stratakis综合征(CSS)、Pacak-zhuang综合征(PZS)等。2007年的研究发现,1886年报道的Fraulein Roll家族后代成员中有4人存在RET原癌基因胚系突变所致的MEN 2。总之,目前认为人类嗜铬细胞瘤发病与遗传易感性密切相关;发病机制主要有“伪缺氧”机制、激酶信号或Wnt信号通路相关等学说。

临床表现
嗜铬细胞瘤可见于任何年龄,但以40至50岁之间好发,发病无明显性别差异。其临床表现主要因肿瘤不适当释放儿茶酚胺所致。儿茶酚胺包括肾上腺素、去甲肾上腺素和多巴胺,是维持人体生理功能的重要神经递质。但嗜铬细胞瘤持续或间断地释放过量的儿茶酚胺则可导致机体功能紊乱。
以往曾据其临床特征,简单地总结为“10%肿瘤”及“6H肿瘤”。
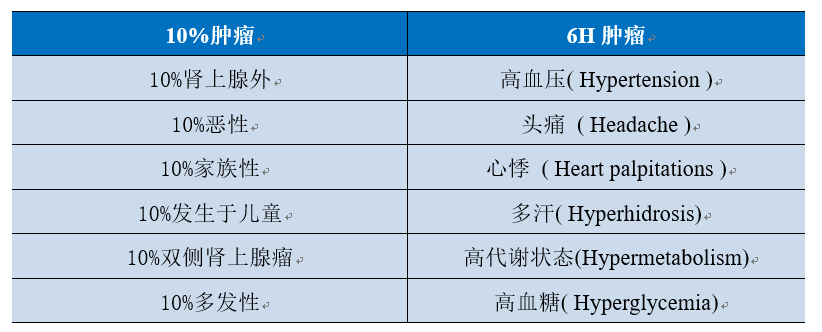
本病最常见的临床表现是高血压。因儿茶酚胺的释放可呈持续性、阵发性,以及少部分肿瘤很少或不合成儿茶酚胺,使患者的血压变化显得“魔幻”:阵发性高血压、持续性高血压、持续性高血压阵发性加剧,或表现为高血压与低血压相交替、甚至出现休克、晕厥等。高血压发作时伴头痛、心悸、多汗三联征对本病的诊断具有重要意义,但出现典型三联征的患者不足25%。某些患者可因长期高血压致严重的心、脑、肾损害或因突发严重的高血压而危及生命,但若能早期获得诊治,则是有望治愈的一种继发性高血压病。
此外,PPGLs患者可因基础代谢增高引起发热、消瘦,因糖代谢紊乱出现高血糖、脂代谢紊乱致血游离脂肪酸增高,少部分病人可有电解质紊乱,如低钾血症、高钙血症。其他可见腹部肿块、消化系统、泌尿系统、血液系统及所伴发遗传性疾病的表现。另有患者可见精神异常,如抑郁或惊恐发作。

诊断
因大部分PPGLs患者的表现较为“魔幻”且临床症状缺乏特异性,一些常规的检查,如心电图、超声检查多无明显异常,甚至许多存在血压升高的患者仅作为高血压病而进行降压治疗,导致本病的误诊、漏诊较多。有研究显示,61%的PPGLs患者以“肾上腺意外瘤”的方式被发现,即在体格检查或检查非肾上腺疾病时经由腹部影像检查而意外发现其肾上腺肿瘤,并最终被证实为PPGLs。 患者的临床表现及家族史等可为诊断PPGLs提供线索,但须依据相关检查作出功能诊断、定位诊断及病因诊断。
1、功能诊断
儿茶酚胺的代谢一般呈“持续性”,释放入血可为“阵发性”,PPGLs肿瘤细胞内的去甲肾上腺素和肾上腺素的中间代谢产物——甲氧基肾上腺素类物质(MNs),包括甲氧基去甲肾上腺素(NMN) 和甲氧基肾上腺素(MN)两种。MNs以“渗漏”方式持续释放入血,最终经肾脏由尿液排泄,或氧化为香草基杏仁酸(VMA)。
最直接的功能诊断为测定血、尿儿茶酚胺,但因儿茶酚胺释放可为“阵发性”,致检查的假阴性率较高。
血浆游离MNs或尿分馏MNs是诊断PPLGs的最佳生化检测指标。
可供临床选用特异性较高的检测项目——测定24小时尿VMA含量。
须注意和尽量避免一些药物对检测结果可能会造成干扰,包括:对乙酰氨基酚、拉贝洛尔、索他洛尔、甲基多巴、三环抗抑郁药、丁螺环酮、酚苄明、单胺氧化酶抑制剂、拟交感药物、可卡因、柳氮磺吡啶和左旋多巴等。
2、定位诊断
B超检查在直径1cm以上的肾上腺肿瘤阳性率较高,因此对于肾上腺及部分肾上腺外(如心脏)肿瘤的定位具有一定的价值。
CT扫描可发现直径1cm以下的肾上腺肿瘤。常规CT平扫无法判断肿瘤有无功能,增强CT检查有助于鉴别肿瘤的性质。
MRI检查无放射性损伤,主要被推荐用于儿童、孕妇、HNPGLs、CT造影剂过敏或存在转移病灶的患者。 化学位移MRI还可鉴别肾上腺腺瘤和其他肾上腺病灶。
但对肿瘤最大径超过5cm、肿瘤分泌去甲肾上腺素或NMN、肾上腺外肿瘤、伴有肿瘤遗传综合征(如SDHB相关)、肿瘤转移风险高以及需功能影像确定病变范围等情况,除了常规的CT或MRI检查,应予功能影像检查。如:18F-FDOPA PET、18F-FDG PET/CT、123IMIBG闪烁扫描以及68Ga-DOTA-SST PET显像等。
3、分子遗传学诊断
PPGLs可伴发遗传综合征。有研究发现11%-13%散发PCCs病例,其实存在着胚系突变。因此,推荐所有PPGLs患者均应接受基因检测,检测的基因至少包括RET、SDHB、SDHD、VHL、NF1和FH基因。MEN1作为可考虑检测的项目。

治疗
目前PPGLs唯一的根治方法仍然是手术治疗,腹腔镜肾上腺手术已被视为治疗肾上腺肿瘤的“金标准”。少数患者,如巨大肿瘤或SDHB突变相关的PCCs(多伴较高的侵袭转移风险)者,采用开放式根治性切除术或许有更多获益。
合理的围术期管理对所有PPGLs患者(包括没有临床症状和高血压的患者)均极为重要。主要包括血压管理、补充血容量及心脏功能的评估。为确定是否已完全摘除肿瘤,推荐术后2-6周查血或尿MNs。若上述生化指标持续异常,则应进一步行影像学检查。
对不能根治性切除的局部肿瘤或远处转移瘤,如果可能应争取减瘤性(姑息性)切除。不能根治切除或仅减瘤性切除的治疗方案,包括放射性核素治疗、CVD联合化疗 (环磷酰胺、长春新碱、氮烯唑胺)、外放射治疗以及针对儿茶酚胺增多的对症处理等。但非手术治疗往往只能提供有限的肿瘤控制和激素水平抑制。
基因分型可用于指导PPGLs的治疗。美国已批准靶向治疗用于携带VHL、RET、BRAF、EPAS1和FGFR1的肿瘤患者,并于2018年7月,正式批准Azedra(iobenguane I 131)注射液,用于治疗PPGLs无法手术切除,且肿瘤已发生扩散,需要系统抗癌治疗的成人和12岁以上青少年患者。预示未来PPGLs将走向基因诊断指导靶向治疗的时代。

随访
尽管大部分PPGLs病人接受手术后可治愈,但在术后5年随访中,仍有5%已完整切除肿瘤的患者发生复发、转移或新发肿瘤。通常的转移部位为局部或远处淋巴结、骨骼、肝脏和肺。目前主张术后所有病人应终身随访,对存在基因突变者应增加随访频次。随访观察内容包括病史与体格检查、血压管理、血或尿MNs,必要时行影像学检查。

展望
在最近的30余年,国内外在PPGLs的临床和分子遗传机制方面的研究取得了许多进展,使人们更好地理解本病病理生理学的路径并对临床处理产生影响,带来了PPGLs诊断治疗的进步。然而,PPGLs患者的自然病史尚未知晓,其预后也不尽相同;疾病的早期诊断、预后判断和转移性肿瘤的治疗等仍然是临床实践中面临的巨大挑战;酪氨酸激酶抑制剂、放射性核素示踪剂、免疫疗法的应用或可使病人获益,但仍需作深入的临床实验观察。
四川大学华西医院于2016年成立“肾上腺疾病诊疗中心”,开展了内分泌科与泌尿外科、放射科、实验医学科、核医学科、病理科等多学科联合会诊(MDT)模式,极大地提高了本病的诊治效率。目前内分泌科肾上腺疾病组正积极开展PPGLs基础与临床研究,以期对本病的分子特征、导致肿瘤发生、转移特定信号通路的改变等进行深入探索。
主要参考文献
[1] Lloyd RV, Osamura RY, Klöppel G, et al. WHO Classification of Tumours of Endocrine Organs[M]. 4th ed. Lyon: IARC Press, 2017.
[2] Jameson JL,Kasper DL,Hartmut P.H.Neumann, et al. Harrison`s Endocrinology[M]. 4th ed. McGraw-Hill Education,2017.
[3] Melmed S, Polonsky KS,Reed Larsen P,et al.Williams Textbook of Endocrinology[M]. 13th ed. Elsevier Inc,2017.
供稿:吴学毅 四川大学华西医院内分泌代谢科








