





戊二酸血症1型(glutaricacidemia type 1,GA1)是一种常染色体隐性遗传病,由于戊二酰辅酶A脱氢酶活性降低或缺失导致赖氨酸、羟赖氨酸及色氨酸分解代谢受阻,代谢产物戊二酰肉碱、戊二酸等在体内异常蓄积,引起代谢紊乱,主要导致神经系统受损。患者临床表现为巨颅、肌张力障碍、运动障碍及发育落后等,常在婴幼儿期由于感染、疫苗接种及手术等诱发急性脑病。由于GA1罕见,临床表现与其他神经系统疾病表现类似,特异性不强,易漏诊或误诊。为了早期诊断和治疗,改善患儿预后,指导临床医师合理诊治,由国内儿科内分泌遗传代谢科专家共同讨论,结合国内外研究进展及国际指南,制定了本共识。
戊二酸血症1型(glutaricacidemia type 1,GA1,OMIM 231670)是一种常染色体隐性遗传病,又称为戊二酸尿症1型(glutaric aciduria type I),是由于戊二酰辅酶A脱氢酶(glutaryl-CoA dehydrogenase,GCDH)活性降低或缺失,导致赖氨酸、羟赖氨酸及色氨酸分解代谢受阻,代谢产物戊二酰肉碱(glutarylcarnitine,C5DC)、戊二酸及3-羟基戊二酸等在体内异常蓄积,引起代谢紊乱,主要导致神经系统受损[1,2,3]。GA1的患病率各地差异较大,国外报道约为1/492 000~1/69 165[4,5],中国约为1/310 200~1/52 078[6,7,8]。GA1临床表现为巨颅、肌张力障碍、运动障碍及发育落后等。常在婴幼儿期由于感染、疫苗接种及手术等诱发急性脑病。由于GA1罕见,临床表现与其他神经系统疾病表现类似,特异性不强,易漏诊或误诊。为了早期诊断和治疗,改善患儿预后,指导临床医师合理诊治,由中国医师协会儿科分会内分泌遗传代谢学组、中华预防医学会出生缺陷预防与控制专业委员会新生儿筛查学组及中华医学会儿科学分会出生缺陷预防和控制专业委员会共同讨论,结合国内外研究进展及国际指南[3],对GA1的诊治达成以下共识。
戊二酸是赖氨酸、羟赖氨酸及色氨酸代谢产物,其代谢通路如图1所示。GCDH是赖氨酸、羟赖氨酸及色氨酸降解通路中的关键酶,参与赖氨酸、羟赖氨酸和色氨酸分解代谢中戊二酰辅酶A脱氢生成3-甲基巴豆酰辅酶A。GCDH基因变异可致GCDH活性降低或丧失,赖氨酸、羟赖氨酸及色氨酸分解代谢受阻,戊二酸、3-羟基戊二酸等代谢产物异常蓄积,并与肉碱结合形成戊二酰肉碱[9]。脑组织中过量的戊二酸及3-羟基戊二酸与兴奋性神经递质谷氨酸结构相似,通过神经递质介导谷氨酸受体过度激活,抑制γ-氨基丁酸合成,抑制性神经递质减少,同时可引起氧化应激反应,造成神经元脱髓鞘、神经元损伤及神经胶质增生[10];另外,戊二酸及3-羟基戊二酸可抑制神经元α-酮戊二酸脱氢酶活性,导致能量障碍和神经元损伤[11]。
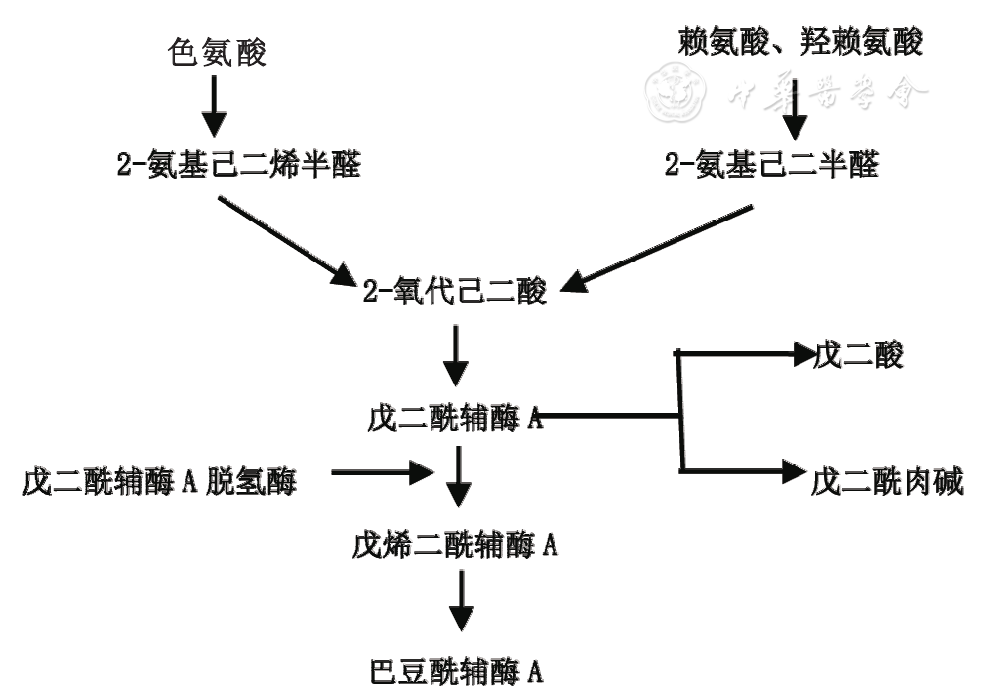
图1 赖氨酸、羟赖氨酸及色氨酸代谢通路图
GA1致病基因GCDH位于染色体19p13.2区,全长约7 kb,包含11个外显子,编码438个氨基酸。GCDH基因变异具有遗传异质性,不同种族和地区常见变异不同,高加索人群最常见的变异为R402W,爱尔兰人群常见变异为E365K[12],非洲血统人常见变异为M405V和V400M[13],中国人常见变异为IVS10-2A>C及c.148T>C (p.W50R)[14,15]。GA1的基因变异类型与生化表型之间存在一定的联系,W50R、E64D、S119L、R128Q、S139L和R402W变异与严重型有关[16]。
2 临床表现
GA1患者常于婴幼儿期发病,临床表现多样,差异较大,以神经系统表现为主,可伴有其他系统异常。未治疗的婴幼儿患者约80%~90%将出现神经系统受累表现,常由感染、发热、疫苗接种及手术等诱发急性脑病发作。
约75%的GA1患者头围增大,部分患儿出生时即有巨颅,部分患儿出生时头围正常,生后不久头围迅速增大[17]。新生儿和婴儿期约50%的患者表现为肌张力低下和运动发育落后,部分患者无症状。患儿常在生后3~36个月发病,出现急性脑病危象,表现为肌张力减退、意识丧失和癫痫发作,恢复期出现进行性肌张力障碍和发育倒退,包括运动、言语、吸吮咀嚼和吞咽反射等[18,19]。随病情进展或急性脑病的复发,神经系统损伤进行性加重,伴认知障碍。未治疗的患者大部分出现基底节(纹状体)损伤,表现为全身肌张力障碍、痉挛性瘫痪、舞蹈症样手足徐动等锥体外系症状,伴发育落后,部分患者智力正常。少部分患者隐匿起病,甚至成年发病,属于晚发型,多表现为非特异性神经系统症状,如头痛、眩晕、共济失调或运动后昏厥、大小便失禁、注意力涣散和感觉异常等,脑白质营养不良多见[20]。
3 实验室检测
常规实验室检测包括血尿常规、肝功能、肾功能、血气分析、血糖、血氨、血乳酸及肌酸激酶等。GA1患者可出现低血糖、血氨及血乳酸升高,代谢性酸中毒、谷丙转氨酶、谷草转氨酶及肌酸激酶升高。
血游离肉碱及酰基肉碱谱水平可通过串联质谱检测,GA1患者血C5DC、C5DC/辛酰基肉碱(capryloylcarnitine,C8)比值(C5DC/C8)、和/或C5DC/丙酰肉碱(propionylcarnitine,C3)比值(C5DC/C3)升高,可伴游离肉碱水平降低。
尿有机酸水平可通过气相色谱质谱技术检测,GA1患者尿戊二酸水平升高,可伴3-羟基戊二酸水平升高。
GCDH基因可通过Sanger测序法及新一代测序技术检测。所有临床疑似患者、质谱检测疑似或诊断的患者及其父母均需要进行基因检测。
4 新生儿遗传代谢病筛查
新生儿遗传代谢病筛查是GA1早期诊断和早期治疗的重要策略,有助于改善预后[23],建议所有新生儿均进行GA1筛查。
新生儿出生48 h后采血,最迟不超过生后7天采血,滴于专用滤纸片上,利用串联质谱检测干血滤纸片中C5DC、C5DC/C8及或C5DC/C3水平进行GA1筛查。
筛查阳性确诊流程见图2。
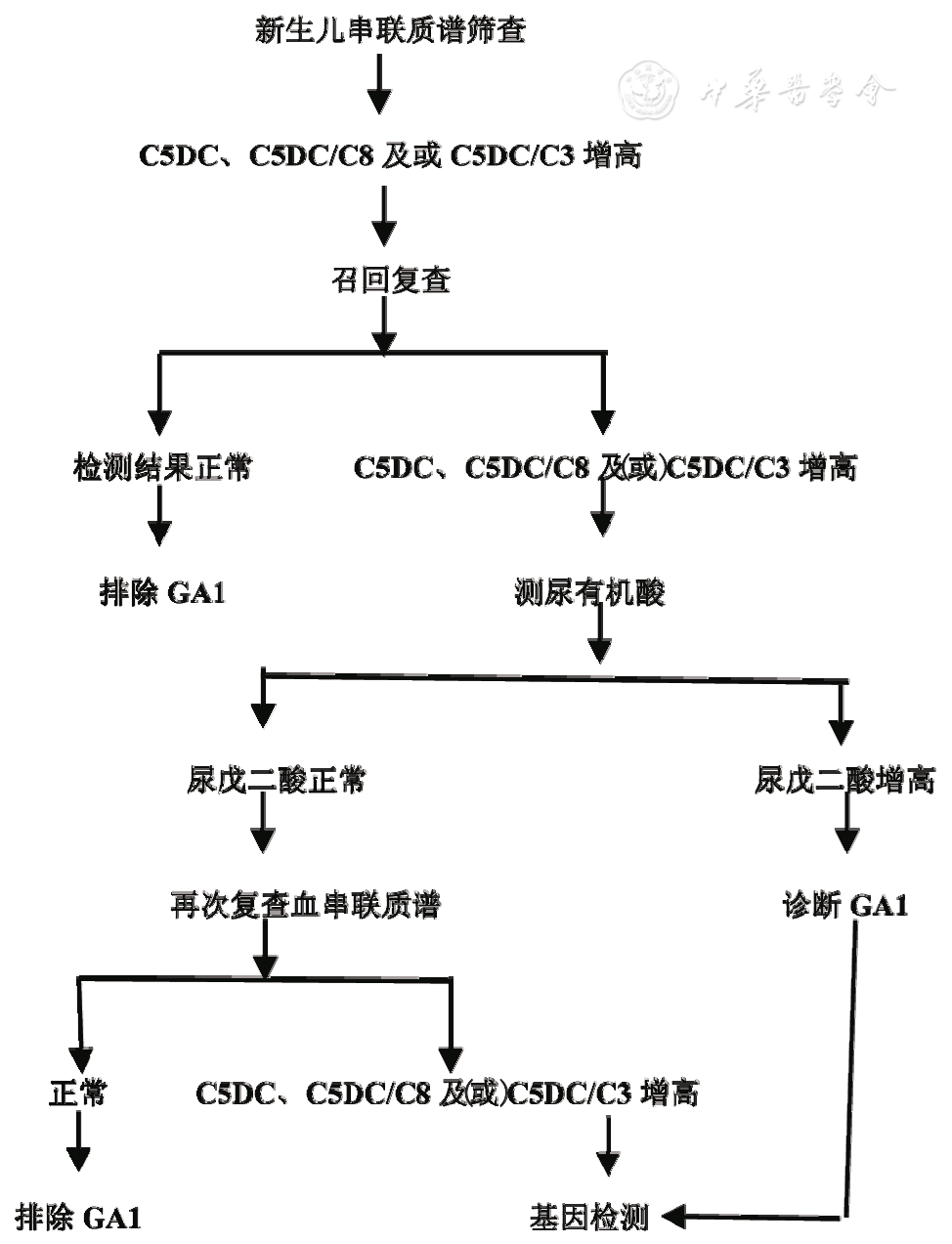
图2 新生儿筛查阳性确诊流程图
筛查假阳性常见于婴儿母亲患GA1或患儿肾功能不全,可导致新生儿血C5DC轻度升高[24],这类患儿建议在肾功能好转后复查血串联质谱。
4.3 新生儿筛查假阴性
由于筛查技术及新生儿个体原因,新生儿筛查存在假阴性[25],故新生儿筛查阴性的患者若出现GA1的相似临床表现,仍需要检测血C5DC及尿戊二酸水平。
5 影像学检查
GA1患者头颅磁共振成像表现多样,典型表现为额颞叶脑实质萎缩,双侧大脑侧裂和颞前极蛛网膜下腔增宽及囊肿,称为"蝶翼状改变",灰质结构中最常见的受累部位为苍白球。20%~30%的GA1患者可出现硬膜下血肿[26]。晚发型GA1患者表现为额颞部发育不全及脑白质营养不良[14,15,16,17,18,19,20,21,22,23,24,25,26,27,28]。
部分患儿脑电图可呈异常放电或癫痫样放电。
孕期超声检查可能发现胎儿双顶径异常增大。
6 诊断
GA1的确诊依据:(1)临床症状:智力、运动及语言发育落后,头围增大。新生儿筛查确诊患者可无临床表现;(2)C5DC、C5DC/C8和(或)C5DC/C3增高;(3)尿戊二酸增高,伴或不伴尿3-羟基戊二酸增高;(4)GCDH基因检测到复合杂合变异或纯合变异。具备(1)、(2)、(3)及(4)条,或(2)、(3)及(4)条即可确诊。具备(1)、(2)及(3)条或(2)及(3)条,可临床诊断,需做GCDH基因检测。
GA1患者婴幼儿期头围增大同时伴进行性脑萎缩、脑室扩大及脑积水,需要与其他原因引起的脑积水及Canavan病引起的头围增大相鉴别;急性脑病危象患者需要与中枢神经系统感染及线粒体脑病鉴别;硬脑膜下腔及蛛网膜下腔出血患者需要与颅脑外伤引起者相鉴别。
又称为戊二酸血症2型。患者临床表现为肝脏、心脏及肌肉病变,很少引起大脑损伤,尿戊二酸水平升高,血C5DC及多种酰基肉碱水平升高,长链酰基肉碱升高显著,急性期常合并低血糖、高脂血症、高尿酸血症等。GA1患者仅C5DC增高,其他酰基肉碱正常,结合基因检测可鉴别。
戊二酸尿症3型是由于琥珀酸-羟甲基戊二酰辅酶辅酶A转移酶(编码基因SUGCT)活性下降或缺乏导致戊二酸转化为戊二酰辅酶A受阻,体内戊二酸累积引起的疾病。临床表现为发育落后及周期性呕吐,急性发作时表现为嗜睡、酮症酸中毒。头颅核磁共振表现为脑白质变性及髓鞘化不良,尿戊二酸显著增高,血C5DC水平正常,故血C5DC水平及基因检测可与GA1相鉴别[28]。
7 治疗
GA1治疗原则:早诊断和早治疗,新生儿筛查确诊的患者即使无症状也应及时治疗,预防或减缓急性脑病危象和神经系统并发症,降低致残率和致死率。治疗方法主要是饮食及左卡尼汀(左旋肉碱)治疗,避免可致急性发作的诱发因素,急性发作时需加强治疗。
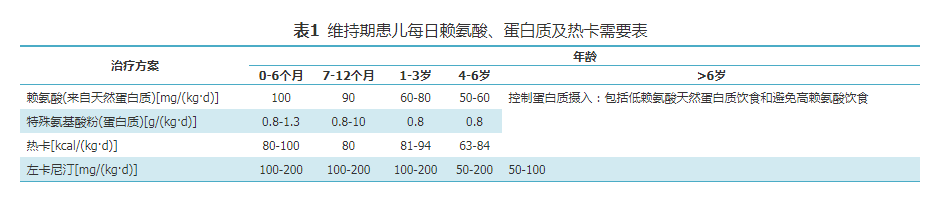
稳定期治疗包括饮食治疗和药物治疗,基本原则是保证患儿正常的生长发育,合理控制代谢产物水平,降低血C5DC、尿戊二酸及3-羟基戊二酸水平,减轻其对神经系统的毒性作用。
7.2 急性期治疗
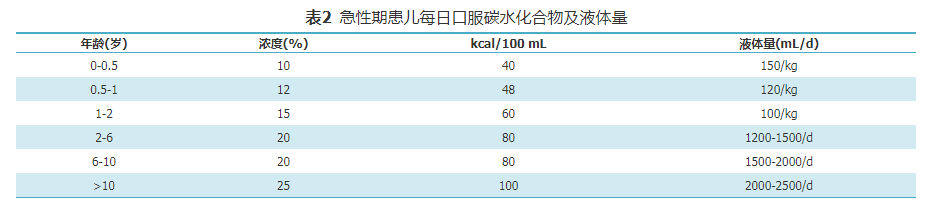
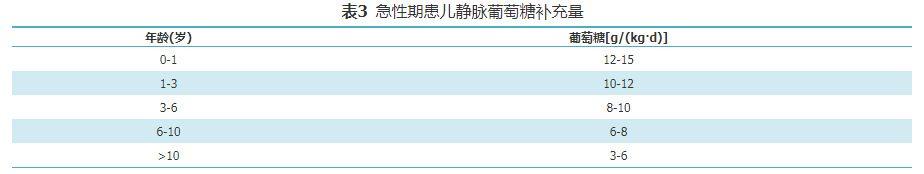
GA1急性发作时,常出现呕吐,腹泻,饮食摄入减少,体温升高或神经系统症状较平时加重,应尽快住院治疗。密切监测心率、血压等生命体征及代谢状况,包括血常规、电解质、血气分析、血乳酸、血氨、肝肾功能、肌酸激酶、血氨基酸和酰基肉碱谱、尿酮体,随时调整治疗方案。具体措施包括:(1)暂停天然蛋白质摄入(最长24 h),在48~72 h内逐步恢复至平时摄入量;(2)增加左卡尼汀剂量,为维持治疗时的2倍,200~300 mg/(kg·d);(3)纠正酸中毒或电解质紊乱;(4)每2 h评估体温、喂养情况及神经系统症状(意识水平、肌张力变化等);(5)可口服碳水化合物,每2 h一次,每日总量见表2,若不能口服,给予静脉补充葡萄糖,用量见表3[3]。若持续空腹高血糖(>8 mmol/L)和(或)糖尿,给予起始剂量0.025~0.05 IU/(kg·h)胰岛素静滴维持血糖在正常范围,防止发生低血糖。
GA1的神经系统并发症主要是运动障碍、硬脑膜下出血、蛛网膜囊肿及癫痫发作,需要联合物理康复、药物和手术治疗。巴氯芬和苯二氮卓类药物(地西泮或氯硝西泮)可作治疗局部和全身肌张力障碍的一线药物。巴氯芬和(或)苯二氮卓类药物治疗无效或出现不良反应时,抗胆碱能药物可作为二线药物[2]。
对于严重肌张力障碍的GA1患者采用立体定向苍白球切开术及深部脑刺激,短期对肌张力有改善,长期效果尚不明确[33]。
需要对患者(年龄较大者)及患者家长进行GA1疾病知识培训,告知平时生活中及免疫接种注意事项。
8 随访监测
GA1患者的定期随访、监测及评估有助于改善预后,降低死亡率。建议多学科团队参与随访监测,包括遗传代谢科医师、普通儿科医师、神经儿科医师、营养师、心理学家、康复师以及社会工作者。监测指标包括发病情况、体格检查、运动及听力发育情况,蛋白质、脂肪及能量摄入量等,1岁内每3个月1次,1~6岁每6个月1次,6岁以后每年1次;头颅MRI可每年检查一次;智力、运动及语言功能每年评估一次;生化监测指标包括游离肉碱、酰基肉碱、尿戊二酸、氨基酸、血常规、肝肾功能及微量元素,1岁内每3个月1次,1~6岁每6个月1次,6岁以后每年1次。
9 预后
GA1患者的预后与首次发病年龄、诊断时间和开始治疗的时间相关。经新生儿疾病筛查,在发病前开始治疗的患者,智商评分可达到平均水平或以上,但精细运动和语言功能仍会受到一定程度的影响。对GA1患儿应开展早期神经心理和语言评估,并给予必要的干预[18]。GA1的预后存在个体差异,即使同胞患者,预后也有较大差异。
10 遗传咨询及产前诊断
GA1属于相对常见的遗传代谢病,对于患者家庭有必要进行遗传咨询及产前诊断。
GA1属于常染色体隐性遗传病,患者家庭成员有患此病的可能性,故对于家庭成员中有发育落后的患者,需要做相关检查,以鉴别是否为GA1患者。另外,患者父母若再生育,所生孩子25%概率患病,50%概率为基因变异携带者,25%概率既不是患者也不是携带者。
方法包括羊水细胞或绒毛组织基因变异检测、羊水代谢物检测及胚胎植入前诊断。
产前诊断GA1患者家系基因变异分析和充分遗传咨询是家庭产期诊断的基础。可对变异明确的患者家系进行产前基因诊断。患者母亲于10~14孕周经绒毛膜穿刺,或16~20孕周经羊水穿刺提取胎儿细胞DNA进行GCDH基因测序。对于基因变异结果不明确,或只检测到一个变异位点的患者母亲,不能进行产前基因诊断。
产前诊断GA1患者母亲在妊娠16~20孕周时,抽取羊水,串联质谱技术检测羊水C5DC水平,计算C5DC/C8比值,气相色谱质谱技术检测羊水戊二酸水平,判断胎儿是否患GA1,具有快速、特异及准确的优点,且可以弥补产前基因诊断的不足[34,35]。建议将基因检测及羊水质谱检测联合用于GA1的产前诊断,提高准确率及效率。
10.2.3 胚胎植入前诊断
利用第三代试管婴儿技术,在胚胎植入前检测胚胎细胞的DNA,进行GCDH基因测序,将未带携GCDH基因2个变异的胚胎植入,并完成产前诊断。
参加本共识制定人员(排名不分先后):上海交通大学医学院附属新华医院(顾学范、韩连书、邱文娟、张惠文);北京大学第一医院(杨艳玲);北京儿童医院(巩纯秀、吴迪);北京市妇幼保健院(孔元原);浙江大学附属儿童医院(赵正言、杨茹莱、黄新文、傅君芬);福建医科大学附属福州儿童医院(陈瑞敏);华中科技大学附属同济医院(罗小平、梁雁、应艳琴);中国人民解放军总医院第七医学中心附属八一儿童医院(封志纯);中国人民解放军总医院第五医学中心南院区(何玺玉);上海交通大学医学院附属儿童医院(李嫔、吕拥芬);复旦大学附属儿科医院(罗飞宏、陆炜);上海交通大学医学院附属上海儿童医学中心(王秀敏);河南省儿童医院(卫海燕、陈永兴);重庆全科大学附属儿童医院(熊丰、朱岷);广州市妇女儿童医疗中心(刘丽、黄永兰);中山大学附属第一医院(马华梅);山东省立医院(李桂梅);济南市妇幼保健院(邹卉);广西医科大学第二附属医院(陈少科、范歆);吉林大学第一院(杜红伟);中国医科大学附属盛京医院(辛颖);哈尔滨医科大学附属第一医院(崔岚魏);苏州大学附属儿童医院(陈临琪);武汉市儿童医院(姚辉);西安交通大学附属儿童医院(汪治华);贵阳市妇幼保健院(刘毓、杨莹);湖南省妇幼保健院(王华);南京市儿童医院(顾威);成都市妇女儿童中心医院(程欣然)
如需原件,可在下方留言并附上邮箱,我们将会邮件回复!








