





10月8日,我们翻译并发布了大分子检测指南 | CLSI C64:蛋白质和肽类的质谱定量第一章,本期为第七章上,文章底部附有往期内容链接,欢迎感兴趣的同道查看。

第七章:方法开发
本章包括
• 从计划到经验优化的方法开发策略
• 方法开发中的实验设计指南(含有推荐的结果)
• 推荐的预验证实验,用于跟踪进展并降低正式的方法验证相关的风险
本章为临床诊断用途的蛋白质和肽分析方法的设计、优化和初步评估提供了指导。在工作的初始阶段进行适当的计划很重要,这样可在项目早期就考虑到机会和局限性。回顾内容应包括临床需求、方法的可行性、技术问题,以及经济效益。图9总结了从计划到初步性能评估所涉及的步骤。尽管图9描述了一个线性的开发过程,但实际上这个过程是迭代的、相互关联的,并且阶段之间有显著的重叠。基于性能评估期间的方法性能,方法开发者可能需要重新优化方法参数、选择新的方法,甚至重新考虑该方法是否能满足最低性能目标。

图9. 方法开发过程总结
7.1 可行性确定和计划
在项目计划阶段,方法开发者应考虑检测的临床效用和预期用途以及检测指征以评估不准确的检测结果可能产生的影响,并确定不准确的检测结果是否对患者构成安全风险。在计划阶段应考虑的技术方面包括开发和常规使用检测方法所需的仪器设备、预期的方法开发和验证时间表、检测通量、所需的周转时间、样本准备时间、仪器分析时间以及自动化的使用。此外,评估实验室实施检测并维持其在常规使用中性能的能力也很重要。通常,方法设计特性会根据现有的实验室仪器设备和基础设施进行实际调整,因为这是方法开发、验证和实施的基础。在计划阶段,开发者应评估财务状况和检测的预期投资回报,并了解所有相关的监管要求。
7.2 被测量的定义
分析策略取决于被测量。因此,收集相关样本类型中分析物所有的结构信息至关重要,包括临床相关的蛋白质形式和分析物的结合物,以便创建测量物的初步定义,并确定测量程序的工作流程。随后,方法开发者应根据被测量的生理浓度、个体间浓度变异、蛋白质形式和临床需求(见第子章节8.1)确定方法的目标性能特征。
7.2.1 结构特性
定义被测量通常从描述不同蛋白质形式的一级、二级、三级和四级结构开始(见第3章)。这种描述通常包括氨基酸序列、PTMs(翻译后修饰)的位置、序列变体(良性或致病性),以及已知的相互作用分子。这些数据可从实验、理论探究(例如,序列鉴定)或者文献中整理得到。许多数据库和生物信息资源有助于特性描述。尽管这些资源很有价值,但用户应该完全理解它们在来源、质量、完整性和归纳整理水平方面的局限性。
文献可能已经充分描述了广泛研究的分析物的结构。对于研究较少的分析物,可能依赖于推断数据(例如,与另一种蛋白质的同源性)。实验鉴定通常是首选,但并非总是可行的。收集到的结构信息有助于方法开发者确定被测量的检测流程,特征性肽段的选择和样本富集策略。
先前发表的蛋白质或肽质谱方法有时可以帮我们省去方法预开发的步骤,但还是推荐进行一定程度的分析物表征以发现任何新的信息以及避免不正确假设的后果。例如,用于检测一个族群的常见蛋白质形式的方法可能无法检测另一族群中常见的序列变体。
7.2.2 计算机模拟分解
对于使用蛋白分解辅助工作流程的方法,有必要确定理想的特征性肽段以便于最好地定义被测量。在计划阶段,使用计算机对蛋白质进行模拟分解。蛋白质或肽段首先以N端至C端单字母氨基酸代码的形式表示,接着使用特异性的蛋白质酶或化学试剂将输入的序列裂解成肽段。计算机模拟分解可以手动进行,也可以通过许多免费软件进行,这些软件通常使用已知的裂解特异性规则或者大规模数据集分析来源的经验规则。
表5. 几种常见的蛋白酶及其裂解特异性
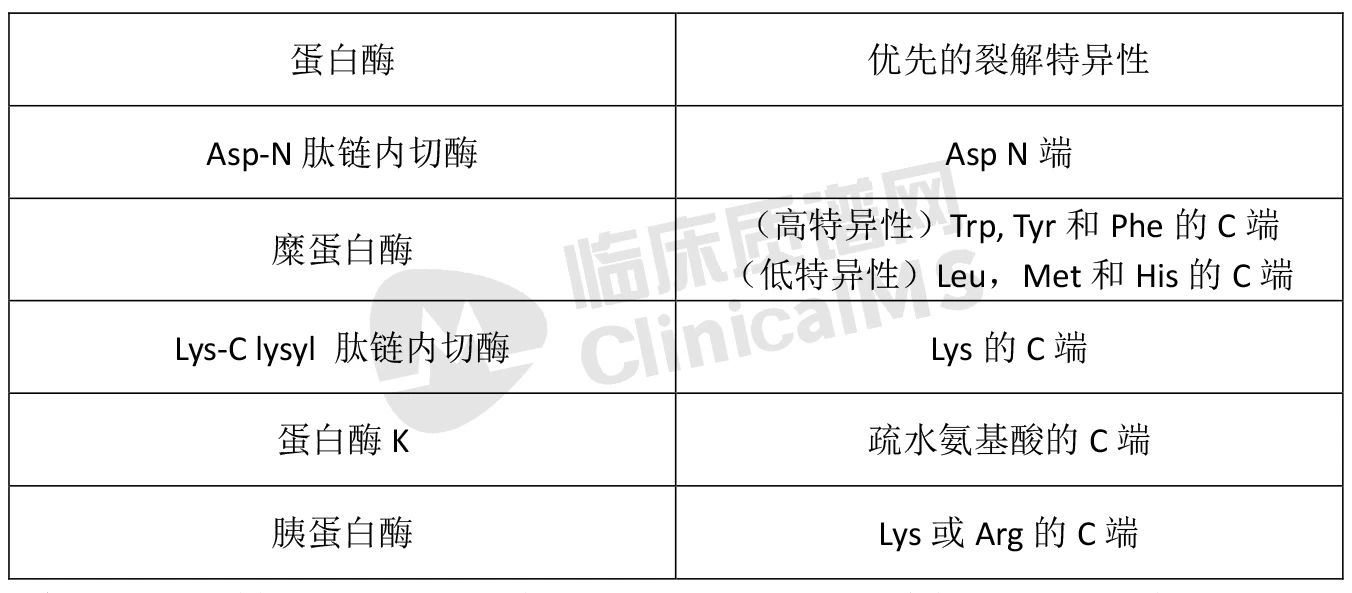
缩写:Arg,精氨酸;Asp,天冬氨酸;His,组胺;Leu,亮氨酸;Lys,赖氨酸;Met,甲硫氨酸;Phe,苯丙氨酸;Trp,色氨酸;Tlyr,酪氨酸。
*对于所述的优先裂解特异性也有例外,例如某些氨基酸(和修饰)以及某些消化条件
实验中,裂解模式可能因多种因素而与预测的分解产物显著不同,包括但不限于体内或体外修饰(例如,广泛的磷酸化妨碍裂解,尿素的羰基化)以及由内源性或外源性蛋白酶产生的N-末端和C-末端。此外,特定的分解条件(例如,pH值、温度)可以影响裂解的速率和特异性。因此,所有预测的或之前报道的分解产物都必须通过实验验证。
7.2.3 序列比对
对于完整蛋白质分析和基于分解的方法,均需要使用序列比对工具将查询的氨基酸序列(例如,完整的蛋白质或特征性肽段)与蛋白质组数据库进行比较。该结果用于评估序列特异性。理想情况下,该序列在被分析的样本中即在人类蛋白质组以及方法引入的任何其他蛋白质组(例如,校准物基质)中是唯一的。
没有任何蛋白质组数据库是全面的。它们通常缺乏对多态性变体和可变剪接变体以及可能在体内发生的蛋白酶水解的注释。因此,序列比对不能为蛋白质或特征性肽段序列的特异性提供最终的证据,但可以表明潜在的特异性的缺乏。即使数据库搜索表明一个序列对于被测量是特异的,也必须实验确认质谱(MS)或串联质谱(MS/MS)检测的特异性。鉴于通过已知的丰度和/或定位差异的分辨(例如,丰富的循环血清蛋白与胞内核转录因子),使用非唯一序列进行方法开发可能是合理的。
7.2.4 候选特征性肽段的选择
对于分解的方法,表6定义了MS分析选择特征性肽段的理想标准。
为实现检测的预期目标可以适当降低标准。例如,一个PTM(翻译后修饰)可能位于比典型的特征性肽段更长的序列中,或者一个小蛋白质可能只产生两个亲水肽。选择指南可以帮助方法开发者将经验性开发工作集中在最符合标准的肽上。同样,当选择非理想肽变得必要时,它们有助于识别潜在的分析挑战。
表6 质谱分析中蛋白水解特征性肽段的选择指南(经牛津大学出版社允许,代表美国临床化学协会,转载自Hoofnagle AN, Whittaker JR, Carr SA, et al. Recommendations for the generation, quantification, storage, and handling of peptides used for mass spectrometry-based assays. Clin Chem. 2016;62(1):48-69.DOI:10.1373/clinchem.2015.250563。)
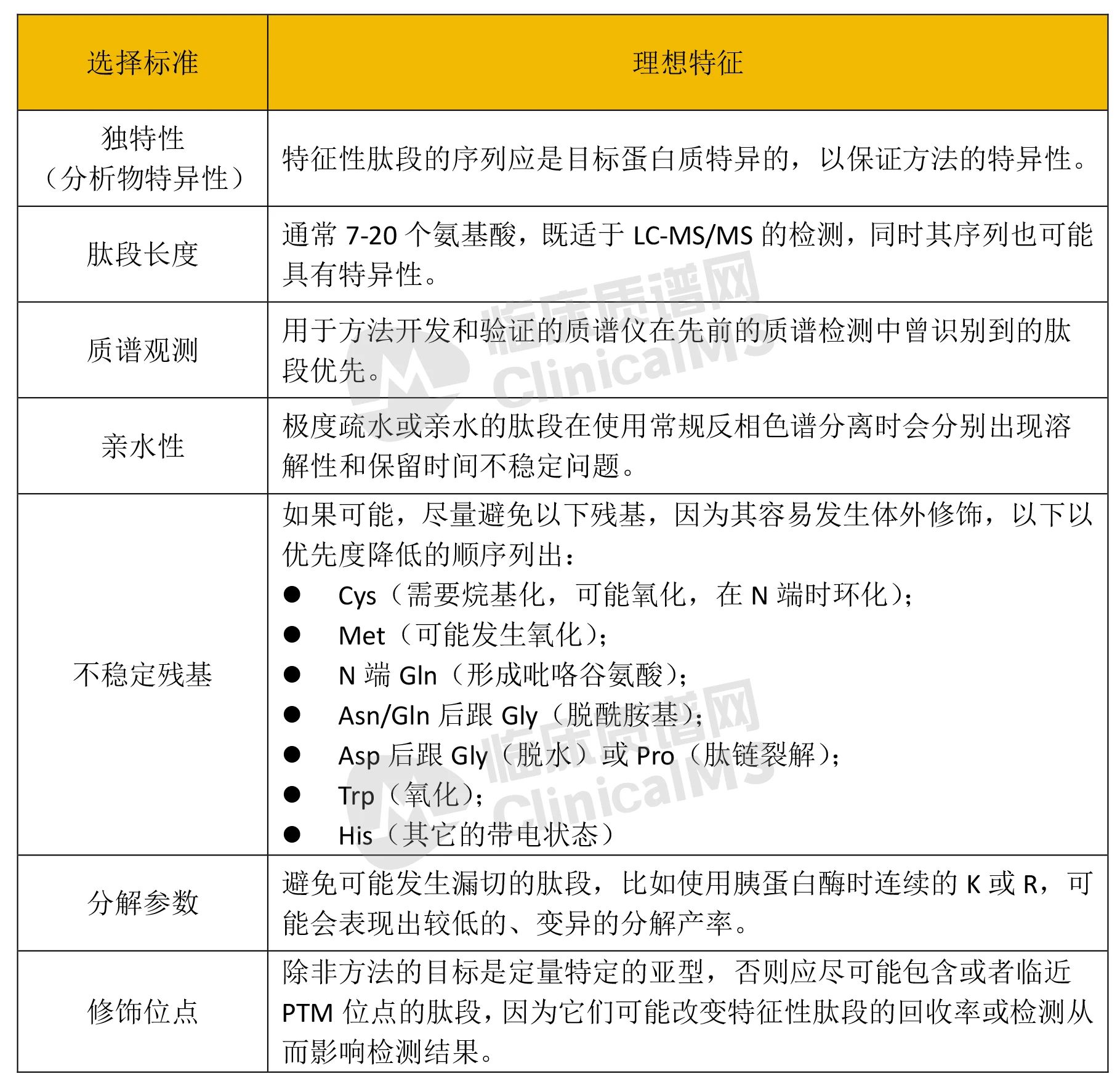
缩写:Asn: 天冬酰胺 (Asparagine),Asp: 天冬氨酸 (Aspartic Acid),Cys: 半胱氨酸 (Cysteine),Gln: 谷氨酰胺 (Glutamine),Gly: 甘氨酸 (Glycine),His: 组氨酸(Histidine),LC-MS/MS: 液相色谱-串联质谱 (Liquid Chromatography-Tandem Mass Spectrometry),Met: 蛋氨酸 (Methionine),MS: 质谱 (Mass Spectrometry), Pro: 脯氨酸 (Proline),PTM: 翻译后修饰 (Post-Translational Modification),RPLC: 反相液相色谱 (Reversed-Phase Liquid Chromatography),Trp: 色氨酸 (Tryptophan)







